加快打造原始創新策源地,加快突破關鍵核心技術,努力搶占科技制高點,為把我國建設成為世界科技強國作出新的更大的貢獻。
——習近平總書記在致中國科學院建院70周年賀信中作出的“兩加快一努力”重要指示要求
面向世界科技前沿、面向經濟主戰場、面向國家重大需求、面向人民生命健康,率先實現科學技術跨越發展,率先建成國家創新人才高地,率先建成國家高水平科技智庫,率先建設國際一流科研機構。
——中國科學院辦院方針



——習近平總書記在致中國科學院建院70周年賀信中作出的“兩加快一努力”重要指示要求
——中國科學院辦院方針




















阿爾茨海默病(AD)是一種尚未被攻克的神經退行性疾病,以Aβ斑塊和tau神經纖維纏結為主要病理特征。隨著我國老齡化加劇,AD患病率持續上升,給社會和家庭帶來沉重經濟負擔。目前AD治療藥物研發進展緩慢。
近年研究發現,小膠質細胞介導的神經炎癥在AD發生發展中起重要作用。小膠質細胞在AD中被Aβ過度持續激活,引發慢性炎癥,加劇tau病理、星形膠質細胞活化及神經元損傷死亡。I型干擾素、TLR信號通路、NLRP3炎癥小體等信號通路被發現參與驅動這一過程,但它們之間的功能聯系尚不明確。
左旋核酸Z-DNA是一種非經典DNA結構,可通過先天免疫受體ZBP1激活免疫反應,在抗病毒及炎癥調控中發揮關鍵作用。左旋核酸配體的鑒定成為領域內的核心科學問題。此外,早在30年前,研究就發現AD患者大腦中存在Z-DNA,但其形成機制、病理功能及ZBP1介導的識別機制的作用仍完全未知。
2025年9月2日,中國科學院上海有機化學研究所生物與化學交叉研究中心許代超研究團隊在Immunity雜志在線發表了題為 "Innate immune sensing of Z-nucleic acids by ZBP1-RIPK1 axis drives neuroinflammation in Alzheimer's disease" 的研究論文,揭示了氧化斷裂的Z型線粒體DNA(Z-mtDNA)可作為ZBP1的新型內源性配體,并驅動小膠質細胞介導的神經炎癥過程。這一發現為理解左旋核酸感知以及AD神經免疫調控機制提供了全新視角。

研究團隊發現,在AD病理環境下,氧化應激導致線粒體DNA(mtDNA)的鳥嘌呤發生氧化形成8-oxoG,隨后DNA斷裂,通過mPTP-VDAC通道釋放至細胞質中。這些斷裂的富含8-oxoG的mtDNA可發生構象轉變,形成非經典的左旋Z型結構(Z-DNA)。這類氧化型Z-DNA可被ZBP1特異性識別,進而通過招募RIPK1并激活其激酶活性,同時促進I型干擾素、TLR信號和NLRP3炎癥小體等多條促炎通路(圖1),最終導致神經炎癥和AD相關病理變化。
研究進一步在AD模型中發現,敲除ZBP1或抑制RIPK1活性可顯著緩解神經炎癥、Aβ沉積和行為缺陷。這一結果不僅確立了Z-DNA–ZBP1–RIPK1軸在AD神經炎癥中的核心作用,也首次闡明氧化應激產生的內源性Z-DNA是激活該通路的關鍵分子。
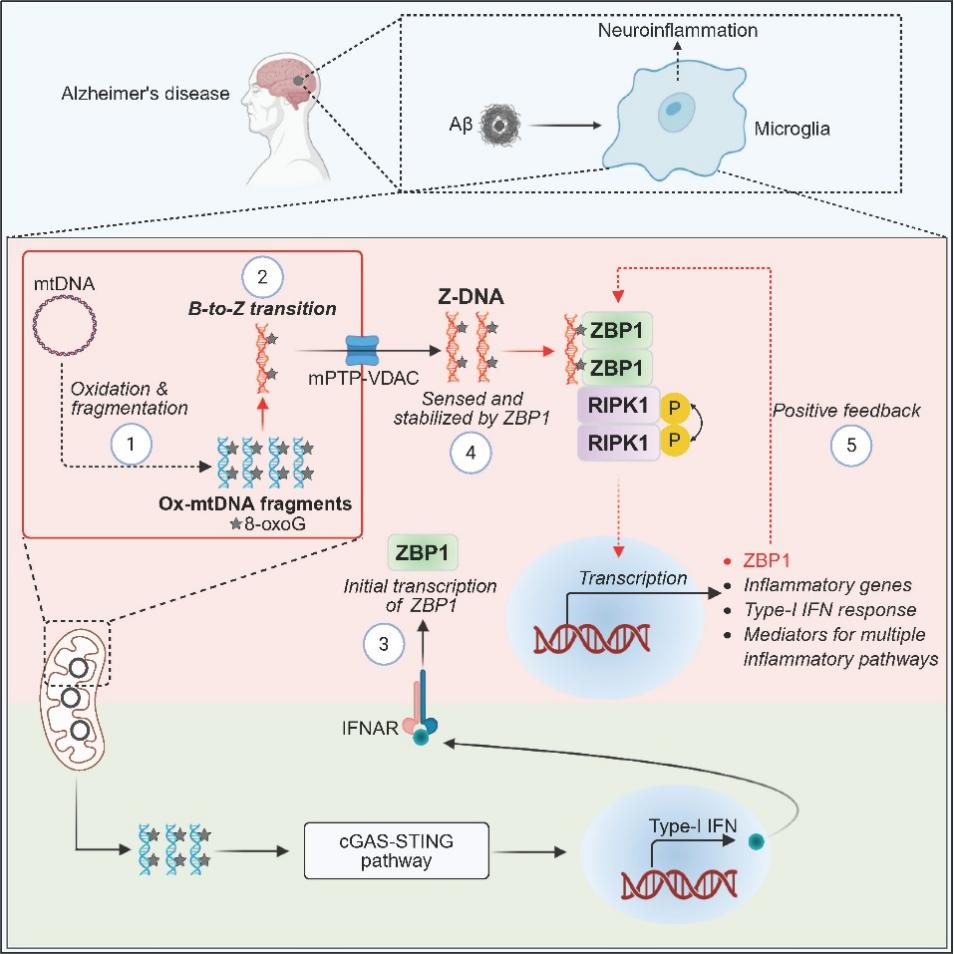
圖1. 新型氧化型左旋核酸的產生及其在AD神經炎癥中的的作用機制模型
總而言之,該研究于鑒定出氧化型左旋核酸作為ZBP1的新型配體,突破了以往認為ZBP1主要識別外源病原體核酸或未修飾內源核酸的認知,同時為AD的免疫機制提供了新視角。由于氧化應激廣泛存在于多種人類疾病當中,該發現也對氧化應激相關的其他人類疾病的機制研究與治療策略具有重要啟示。目前RIPK1抑制劑已處于臨床試驗階段,本研究為其應用于AD治療提供了理論依據,未來或可與Aβ靶向治療聯合應用,為AD患者帶來新的治療希望。
中國科學院生物與化學交叉研究中心許代超研究員為本文通訊作者,浙江大學良渚實驗室莫瑋教授和蘇州大學張健副教授為本文共同通訊作者;中國科學院生物與化學交叉研究中心博士生宋子雯為第一作者,博士后解醒醒和博士生陳宇璐為共同第一作者。該研究獲得了科技部“科技創新-2030腦科學”重大研究計劃,基金委青年基金(A類)、重大研究計劃,上海市“尚思探索學者”、科技先行區、市級重大專項,以及中國科學院相關計劃等項目的支持。
原文鏈接:https://doi.org/10.1016/j.immuni.2025.07.024







 日韩欧美精品三级,激情婷婷亚洲,日韩午夜一区,夜夜夜精品看看
主站蜘蛛池模板:
国产剧情在线观看一区|
久久一区91|
激情欧美一区二区三区在线观看|
亚洲欧美精品在线观看|
91精品久久久久久久久|
亚洲不卡一区二区三区|
亚洲精品720p|
亚洲一区在线电影|
蜜桃视频欧美|
国产精品自拍小视频|
国产一区二区三区中文|
精品一区二区三区在线视频|
欧美日韩精品一区二区三区蜜桃|
欧美色女视频|
欧美一二三视频|
亚洲综合不卡|
久久久久久com|
日韩免费av|
国产精品乱码|
欧美激情黄色片|
婷婷中文字幕一区|
午夜精品一区二区三区视频免费看
|
国产精品区免费视频|
久久99精品久久久久久久久久久久|
国产99久久|
俺去亚洲欧洲欧美日韩|
久久视频一区二区|
久久久久久国产精品免费免费|
亚洲女同另类|
国产真实乱偷精品视频免
|
国产亚洲视频在线观看|
成人羞羞网站|
亚州国产精品久久久|
最新国产拍偷乱拍精品|
北条麻妃国产九九九精品小说|
99精品国产热久久91蜜凸|
蜜桃av一区二区三区电影|
欧美日韩激情一区二区三区|
欧美激情视频一区二区三区在线播放
|
国产二区精品|
国产成人aaaa|
日韩欧美精品三级,激情婷婷亚洲,日韩午夜一区,夜夜夜精品看看
主站蜘蛛池模板:
国产剧情在线观看一区|
久久一区91|
激情欧美一区二区三区在线观看|
亚洲欧美精品在线观看|
91精品久久久久久久久|
亚洲不卡一区二区三区|
亚洲精品720p|
亚洲一区在线电影|
蜜桃视频欧美|
国产精品自拍小视频|
国产一区二区三区中文|
精品一区二区三区在线视频|
欧美日韩精品一区二区三区蜜桃|
欧美色女视频|
欧美一二三视频|
亚洲综合不卡|
久久久久久com|
日韩免费av|
国产精品乱码|
欧美激情黄色片|
婷婷中文字幕一区|
午夜精品一区二区三区视频免费看
|
国产精品区免费视频|
久久99精品久久久久久久久久久久|
国产99久久|
俺去亚洲欧洲欧美日韩|
久久视频一区二区|
久久久久久国产精品免费免费|
亚洲女同另类|
国产真实乱偷精品视频免
|
国产亚洲视频在线观看|
成人羞羞网站|
亚州国产精品久久久|
最新国产拍偷乱拍精品|
北条麻妃国产九九九精品小说|
99精品国产热久久91蜜凸|
蜜桃av一区二区三区电影|
欧美日韩激情一区二区三区|
欧美激情视频一区二区三区在线播放
|
国产二区精品|
国产成人aaaa|
